


亮点评述
化工学报|双金属Co/Zn-ZIFs中C3H6和C3H8吸附和扩散行为分子模拟研究
双金属Co/Zn-ZIFs中C3H6和C3H8吸附和扩散行为分子模拟研究
齐昊 1  王玉杰 1,2 李申辉 1邹琦 2刘轶群 2赵之平 1
王玉杰 1,2 李申辉 1邹琦 2刘轶群 2赵之平 1
(1. 北京理工大学化学与化工学院,北京 102488; 2. 中石化(北京)化工研究院有限公司,北京 100013 )
DOI:10.11949/0438-1157.20241314
引用本文:齐昊, 王玉杰, 李申辉, 邹琦, 刘轶群, 赵之平. 双金属Co/Zn-ZIFs中C3H6和C3H8吸附和扩散行为分子模拟研究[J]. 化工学报, 2025, 76(5): 2313-2326(QI Hao, WANG Yujie, LI Shenhui, ZOU Qi, LIU Yiqun, ZHAO Zhiping. Molecular simulation study on adsorption and diffusion of C3H6 and C3H8 on Co/Zn-ZIFs[J]. CIESC Journal, 2025, 76(5): 2313-2326)
引 言
1 模拟方法
1.1 双金属Co/Zn-ZIFs分子模型的构建
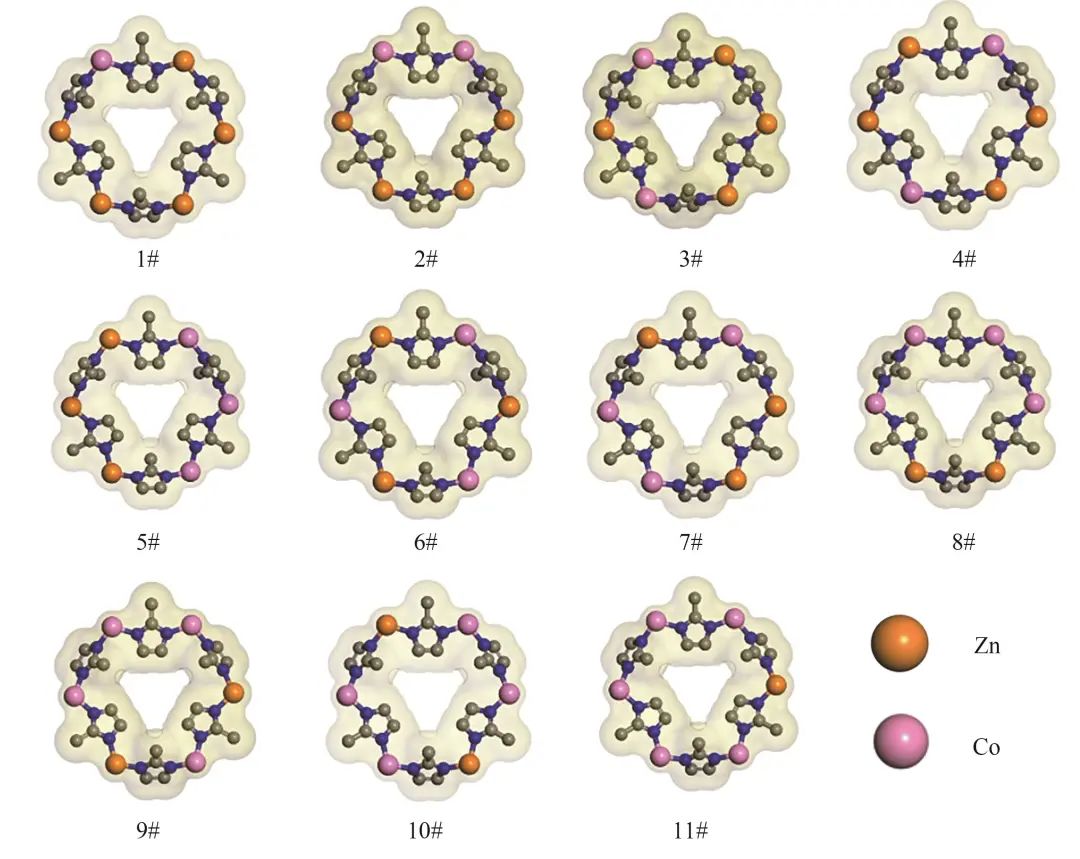
1.2 Zn/Co-ZIFs分子模型点电荷的计算
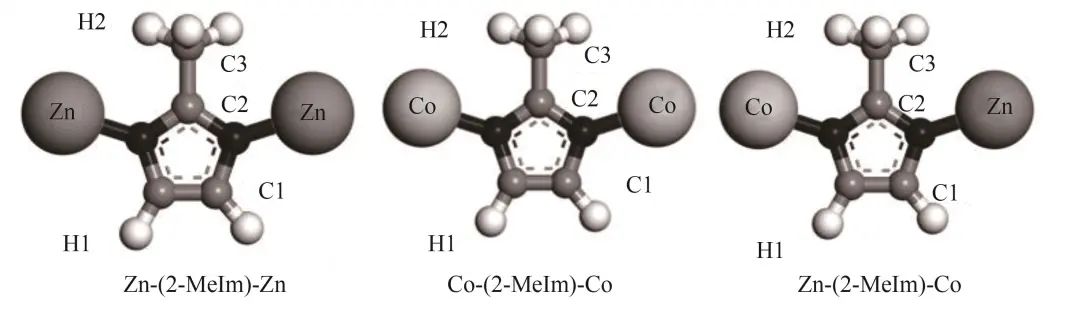
表1 Co/Zn-ZIFs分子模型的点电荷Table 1 Point charges for molecular modeling of Co/Zn-ZIFs
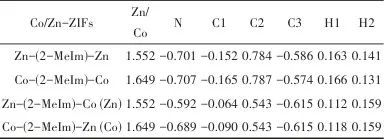
注:点电荷的单位为C。
1.3 气体分子在Co/Zn-ZIFs中吸附特性计算方法
表2 TraPPE力场中C3H8和C3H6的LJ参数Table 2 Lennard-Jones parameters for propane and propylene in the TraPPE force field
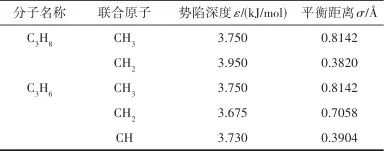
1.4 气体分子在Co/Zn-ZIFs窗口处扩散势垒计算方法
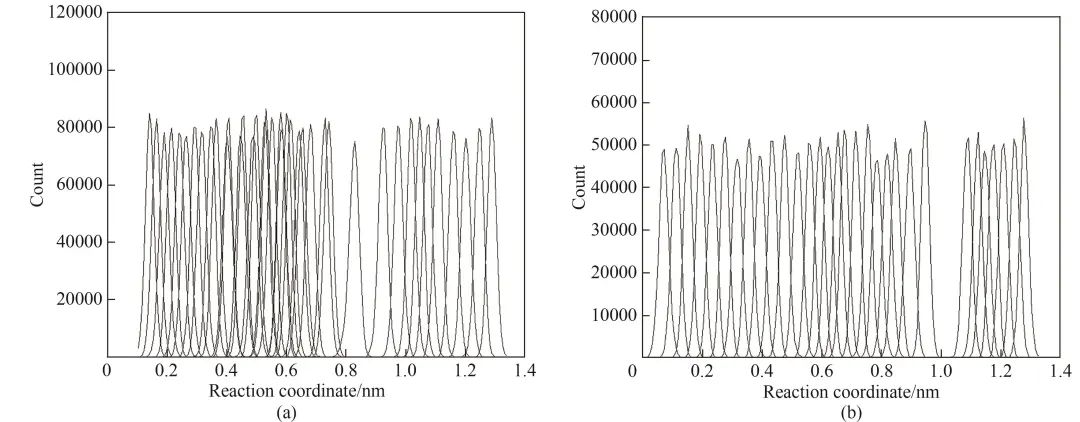
1.5 扩散系数及扩散选择性计算
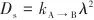 | (1) |
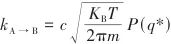 | (2) |
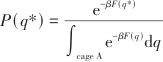 | (3) |
2 结果与讨论
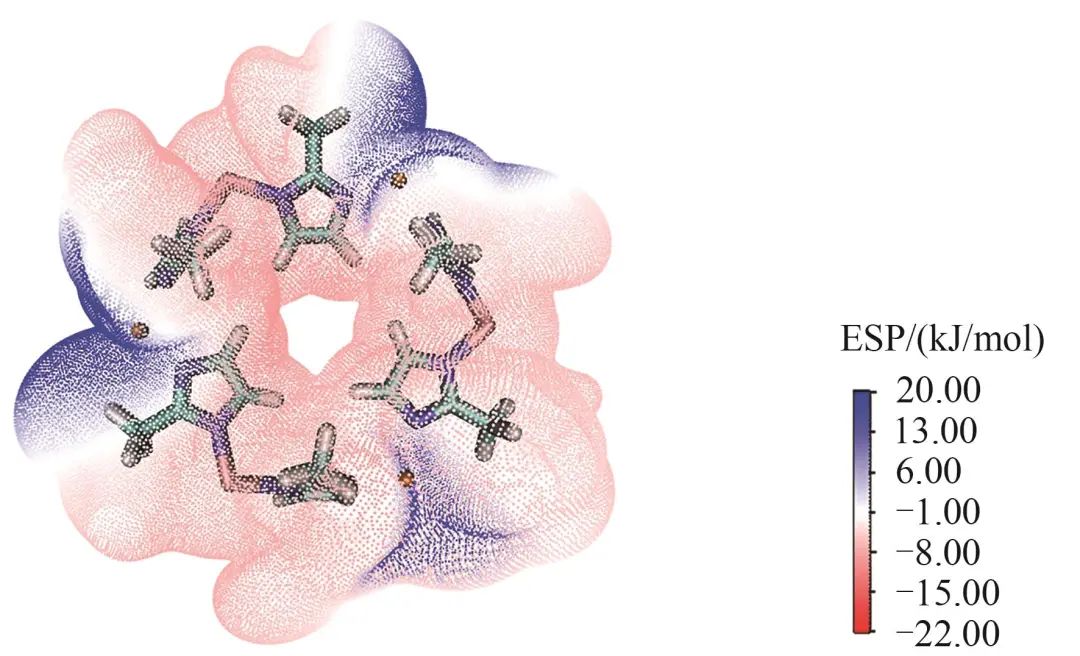
2.1 C3H6和C3H8分子在Co/Zn-ZIFs中吸附特性
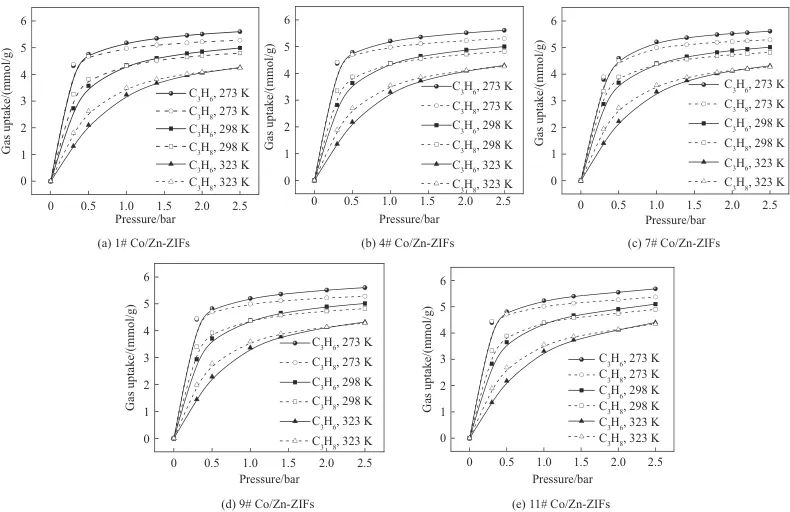
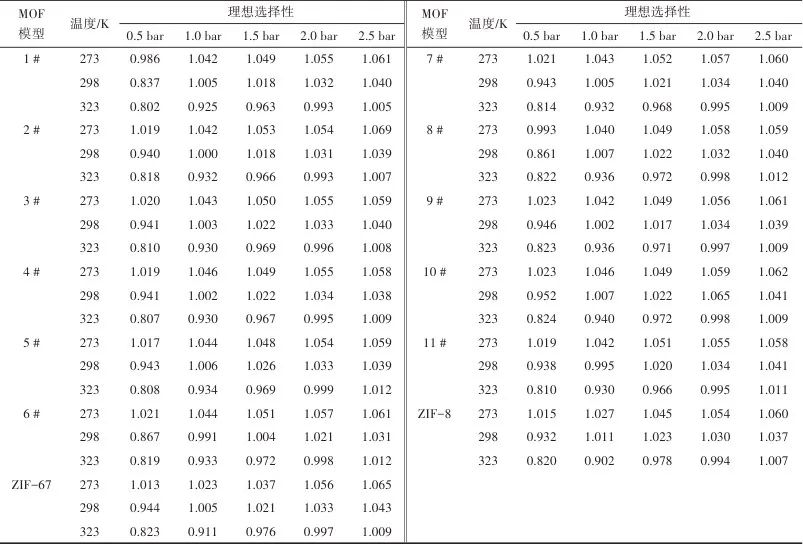
表3 不同温度压力下不同金属比例Co/Zn-ZIFs中C3H6/ C3H8理想选择性Table 3 Ideal selectivity of C3H6/ C3H8 in Co/Zn-ZIFs with different metal ratios at different temperatures and pressures
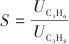 | (4) |
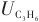 、
、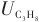 分别为一定温度、压力下,纯C3H6、纯C3H8在Co/Zn-ZIFs上的吸附量。
分别为一定温度、压力下,纯C3H6、纯C3H8在Co/Zn-ZIFs上的吸附量。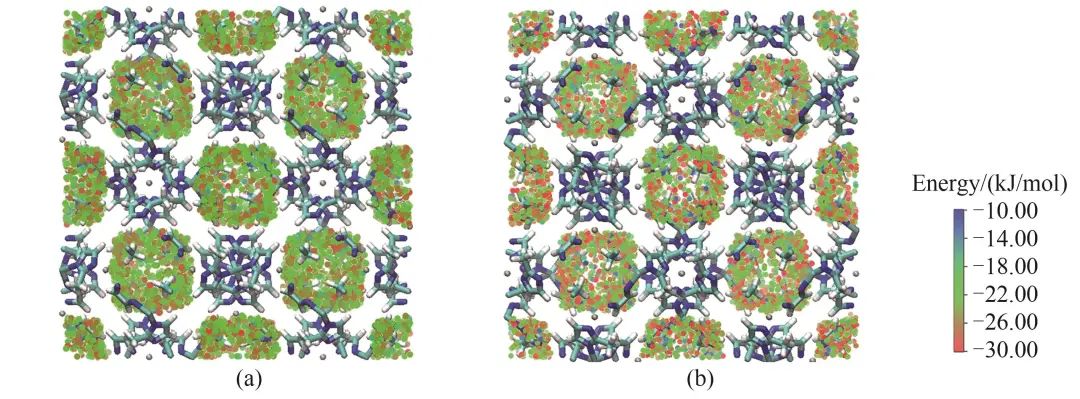
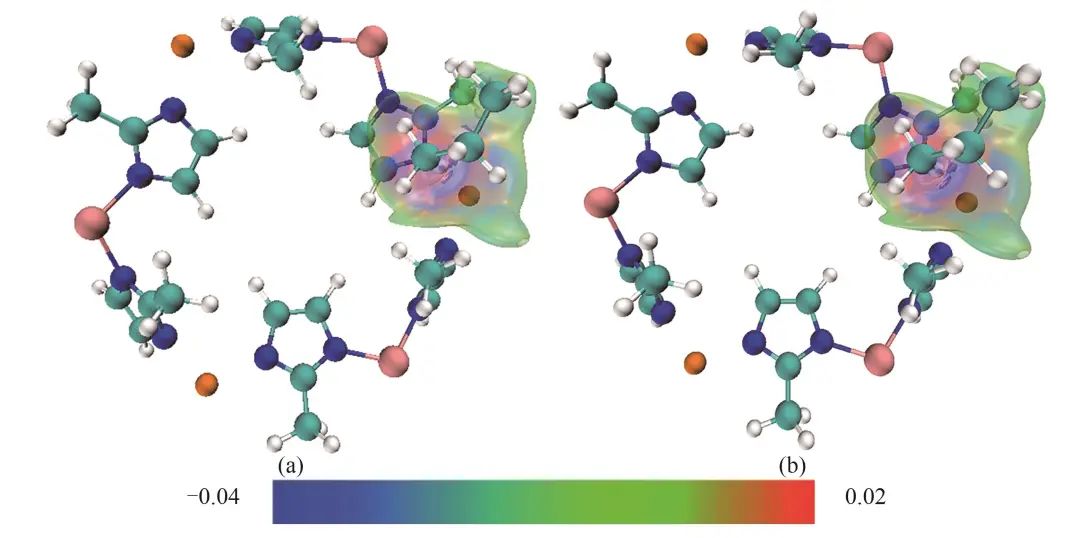
 | (5) |
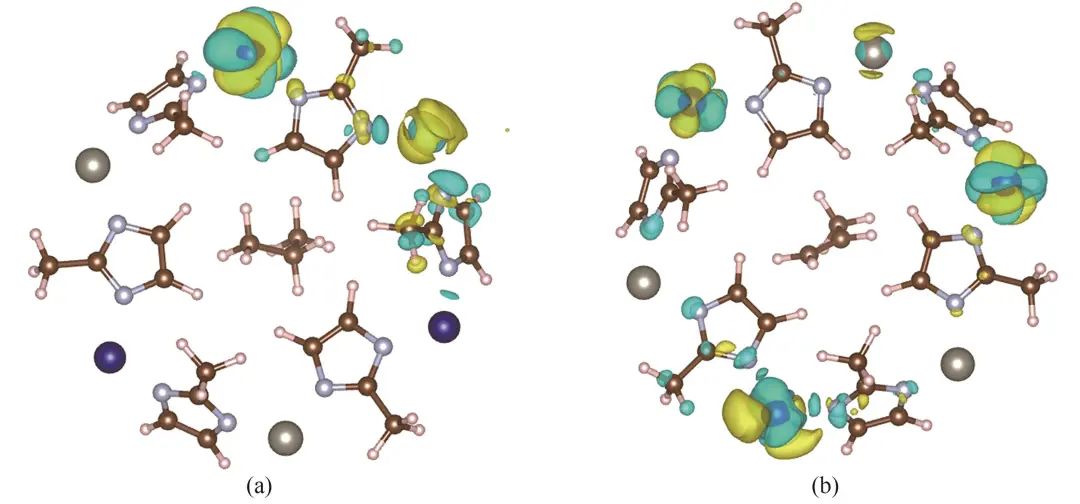
表4 298 K, 10-5 bar下Co/Zn-ZIFs的吸附热Table 4 Adsorption heat of Co/Zn-ZIFs at 298 K, 10-5 bar
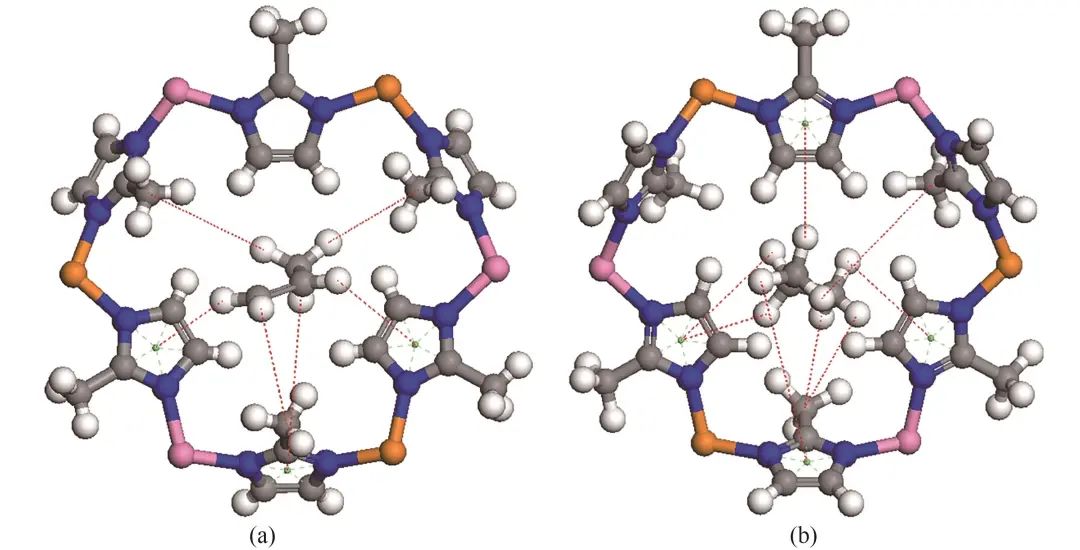
2.2 C3H6和C3H8分子在Co/Zn-ZIFs中扩散特性
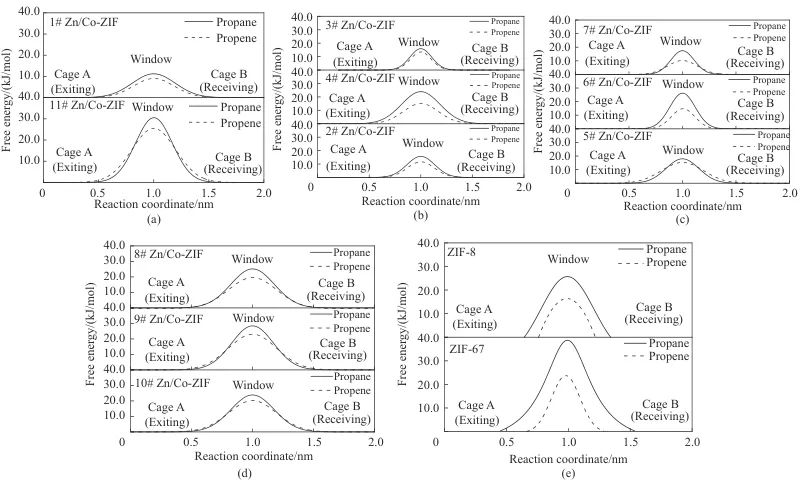
表5 不同金属比例下C3H6、C3H8在Co/Zn-ZIFs中的扩散自由能Table 5 Diffusion free energy of C3H6 and C3H8 in Co/Zn-ZIFs with different metal ratios
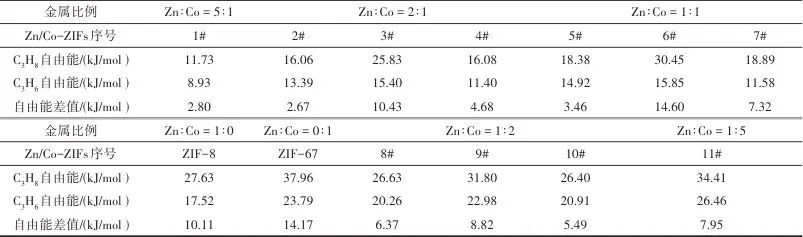
表6 不同Co/Zn-ZIFs分子模型中C3H6、C3H8自扩散系数Table 6 Self-diffusion coefficients of C3H6 and C3H8 in different Co/Zn-ZIFs molecular models
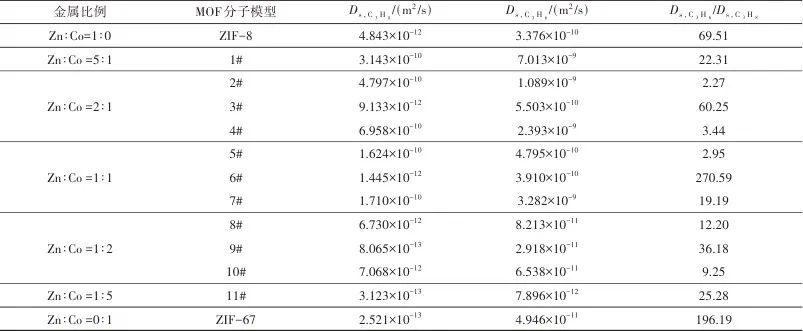
3 结 论

Molecular simulation study on adsorption and diffusion of C3H6 and C3H8 on Co/Zn-ZIFs
QI Hao 1 WANG Yujie 1,2 LI Shenhui 1ZOU Qi 2LIU Yiqun 2ZHAO Zhiping 1
(1. School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, China; 2. Sinopec Beijing Research Institute of Chemical Industry, Beijing 100013, China )

第一作者:齐昊(1998—),男,硕士研究生,2765618801@qq.com
通讯作者:王玉杰(1980—),女,博士,高级工程师,wangyuji.bjhy@sinopec.com;赵之平(1963—),男,博士,教授,zhaozp@bit.edu.cn



